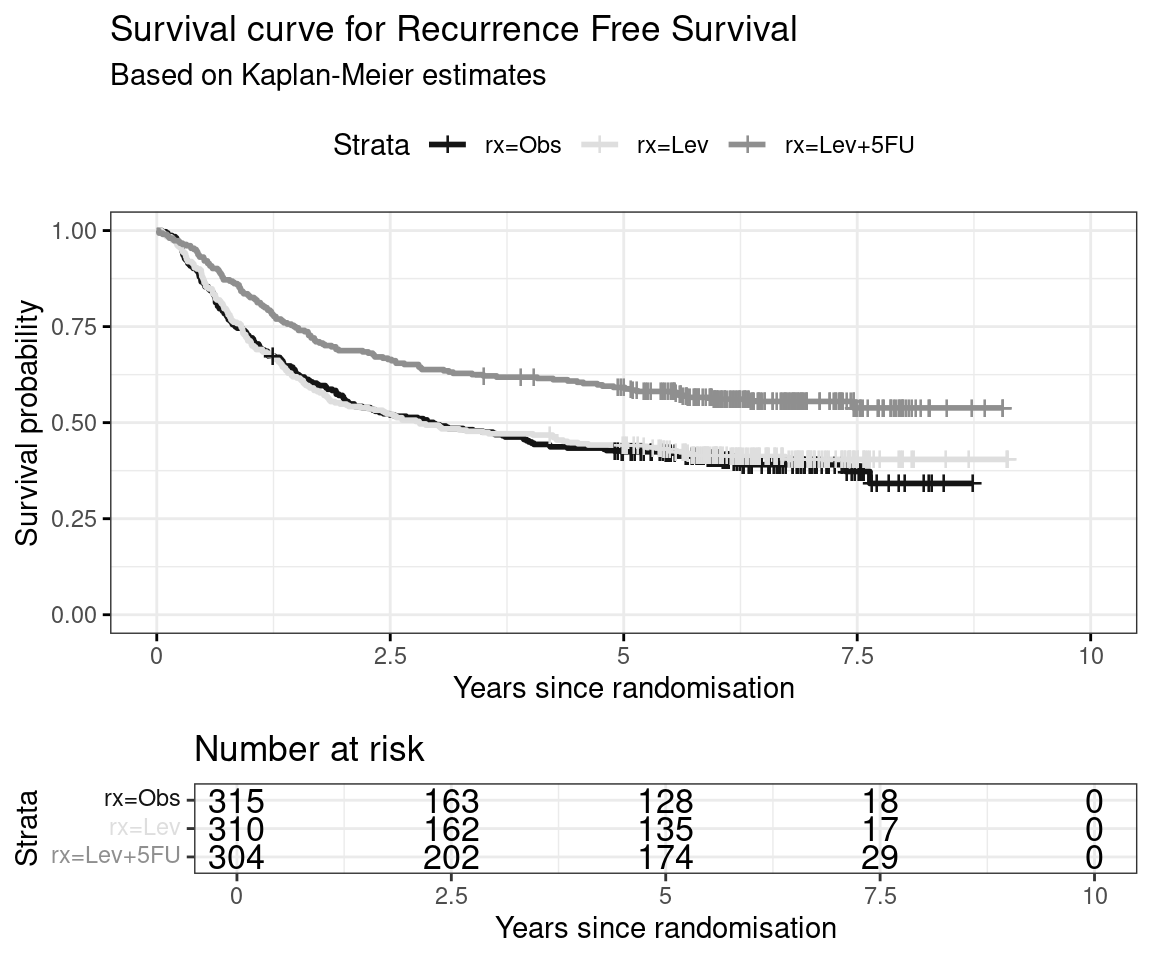
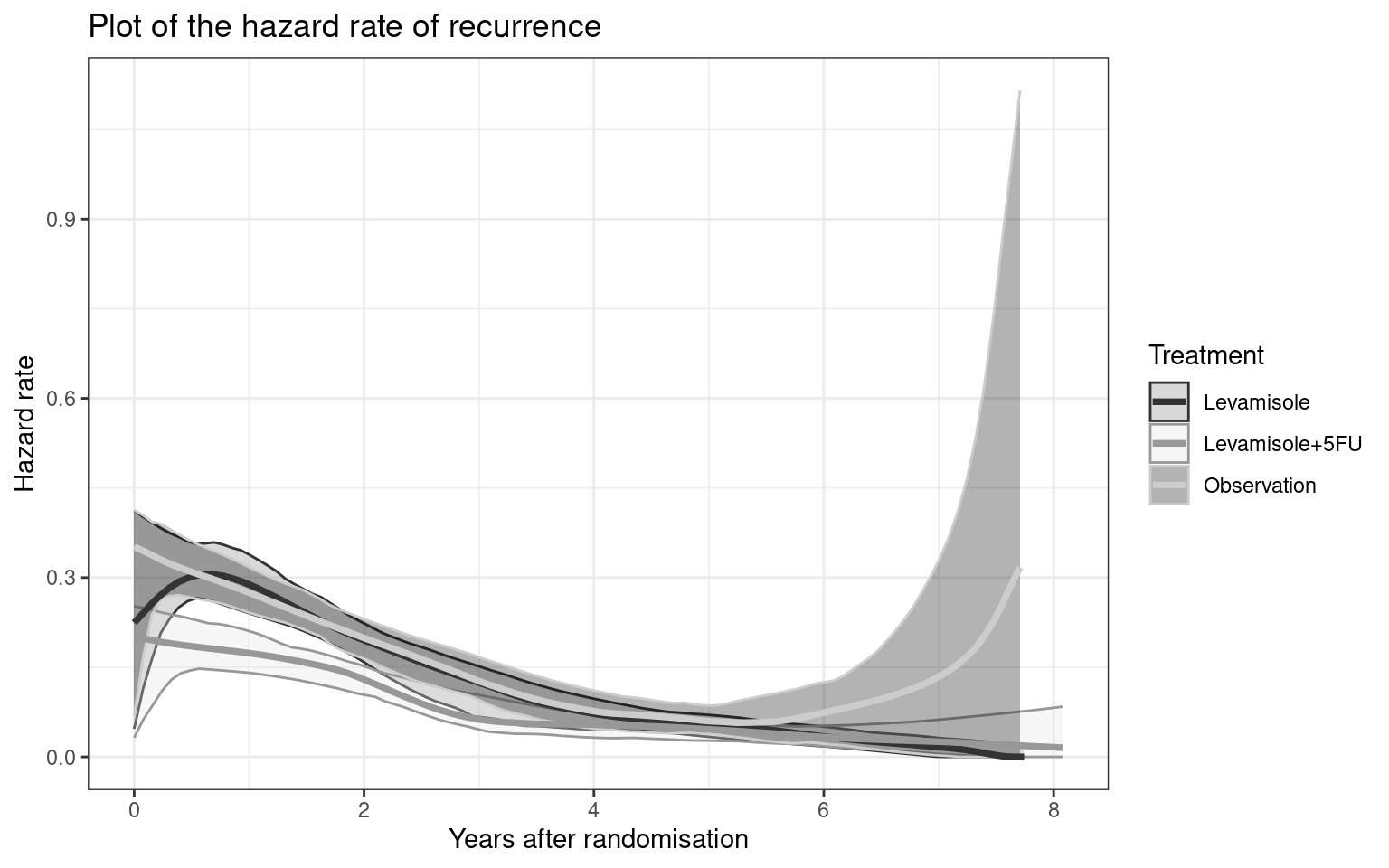
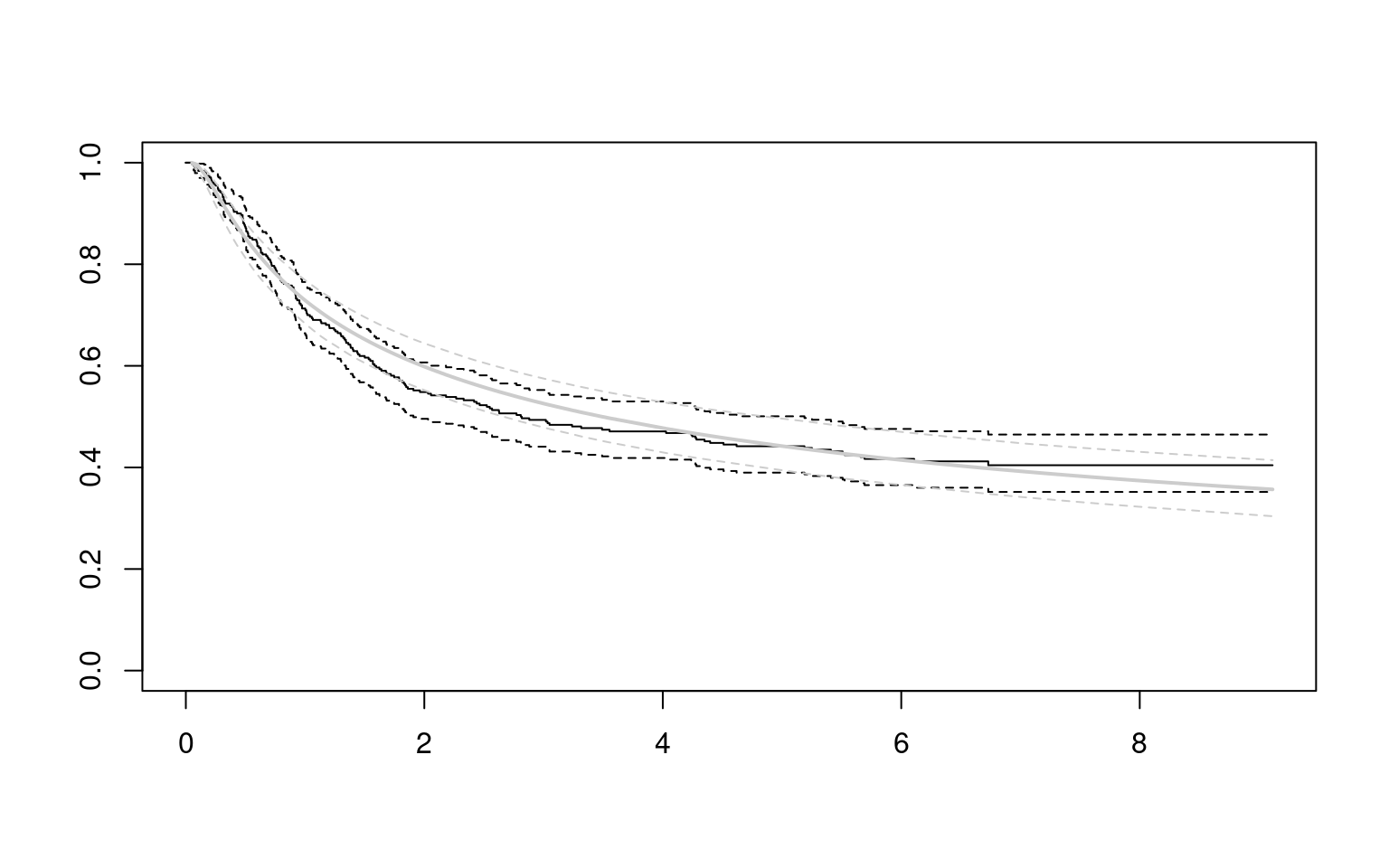
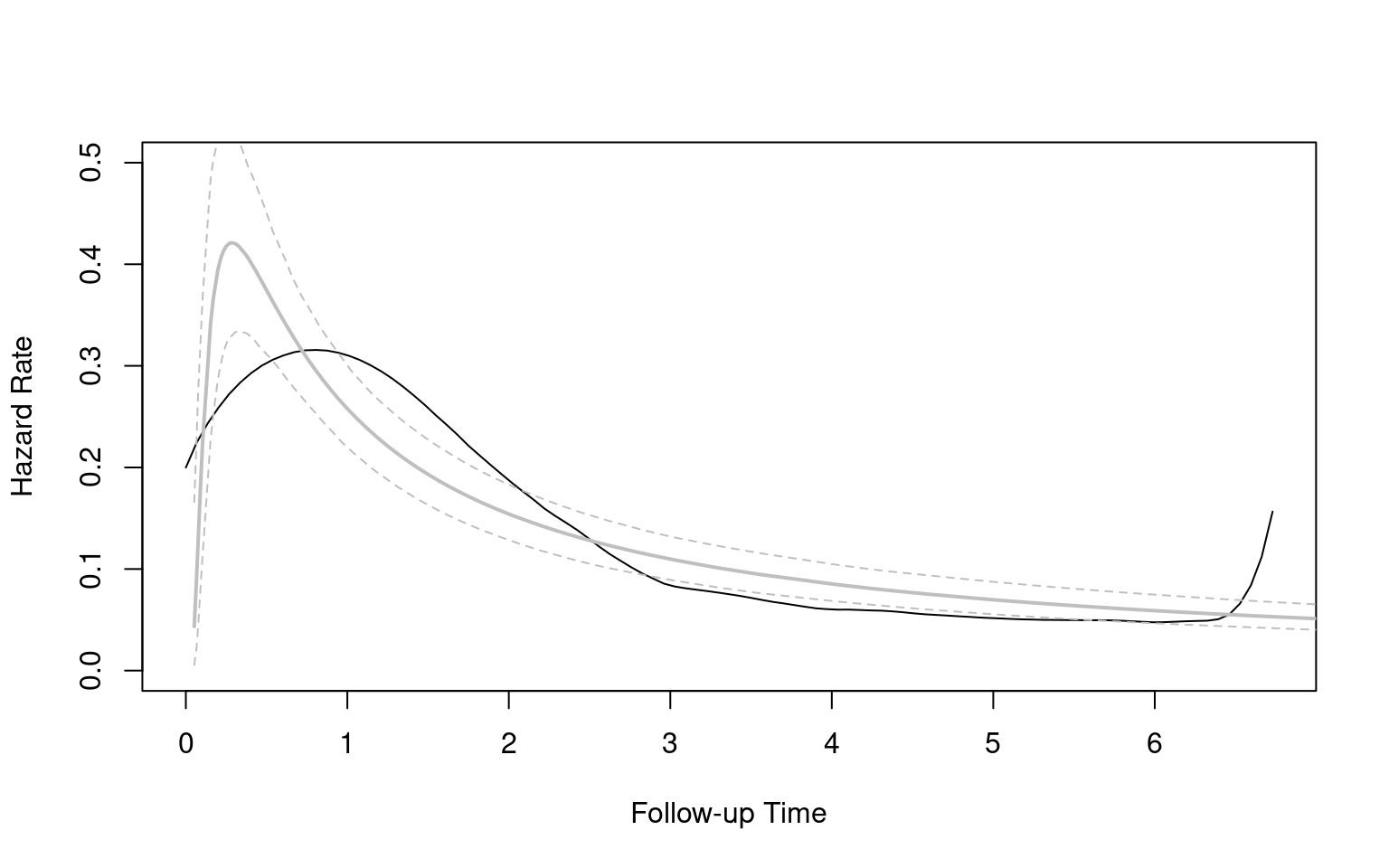
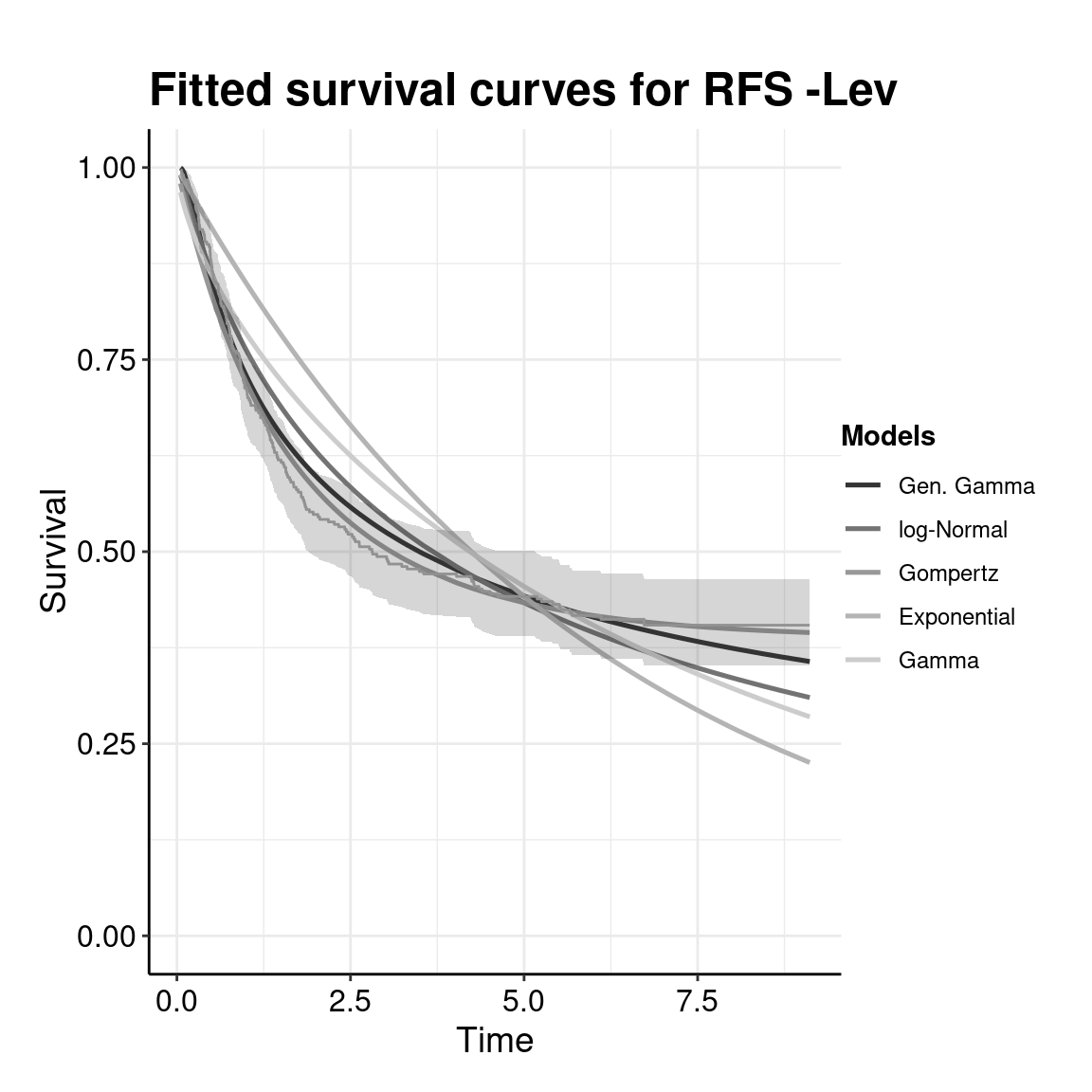
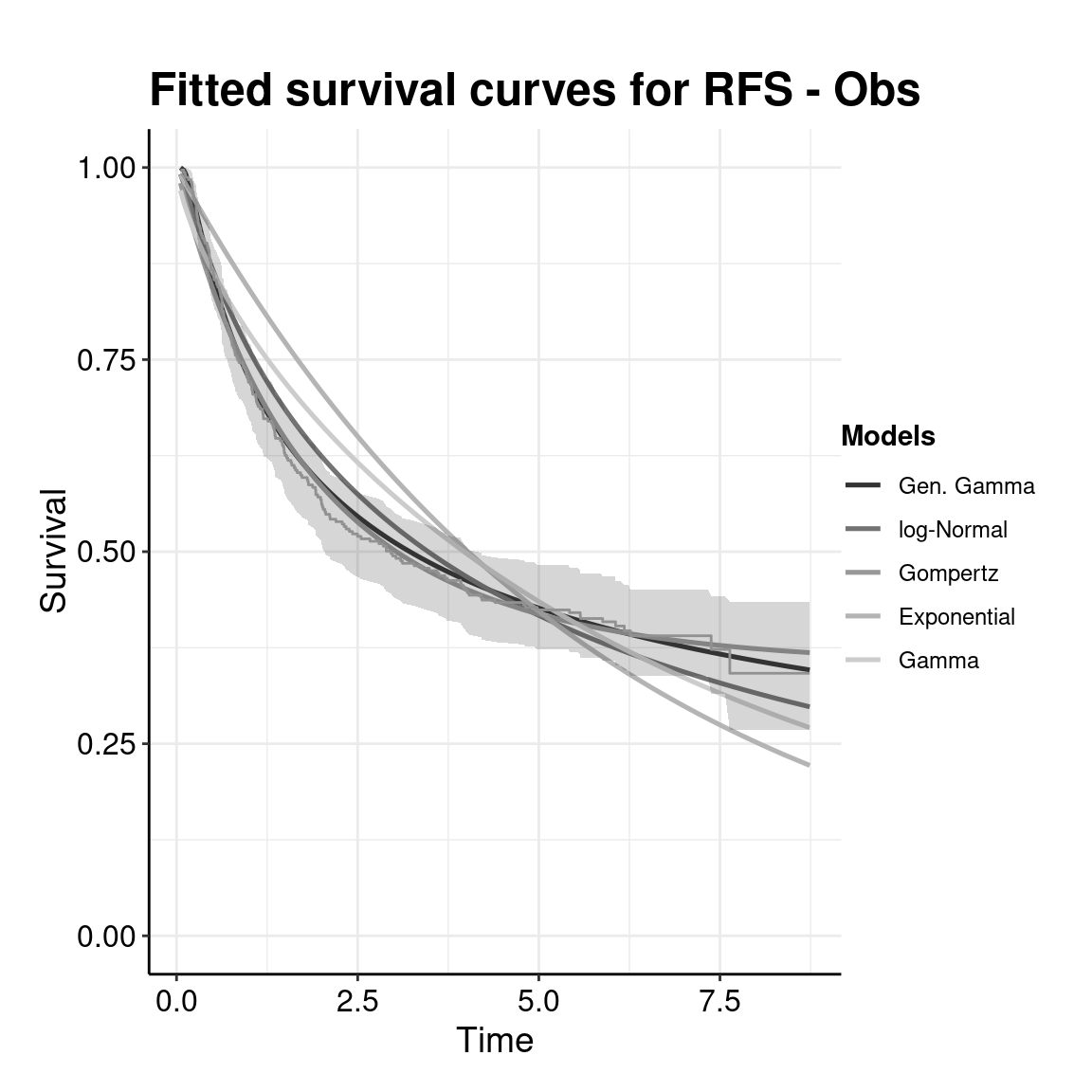
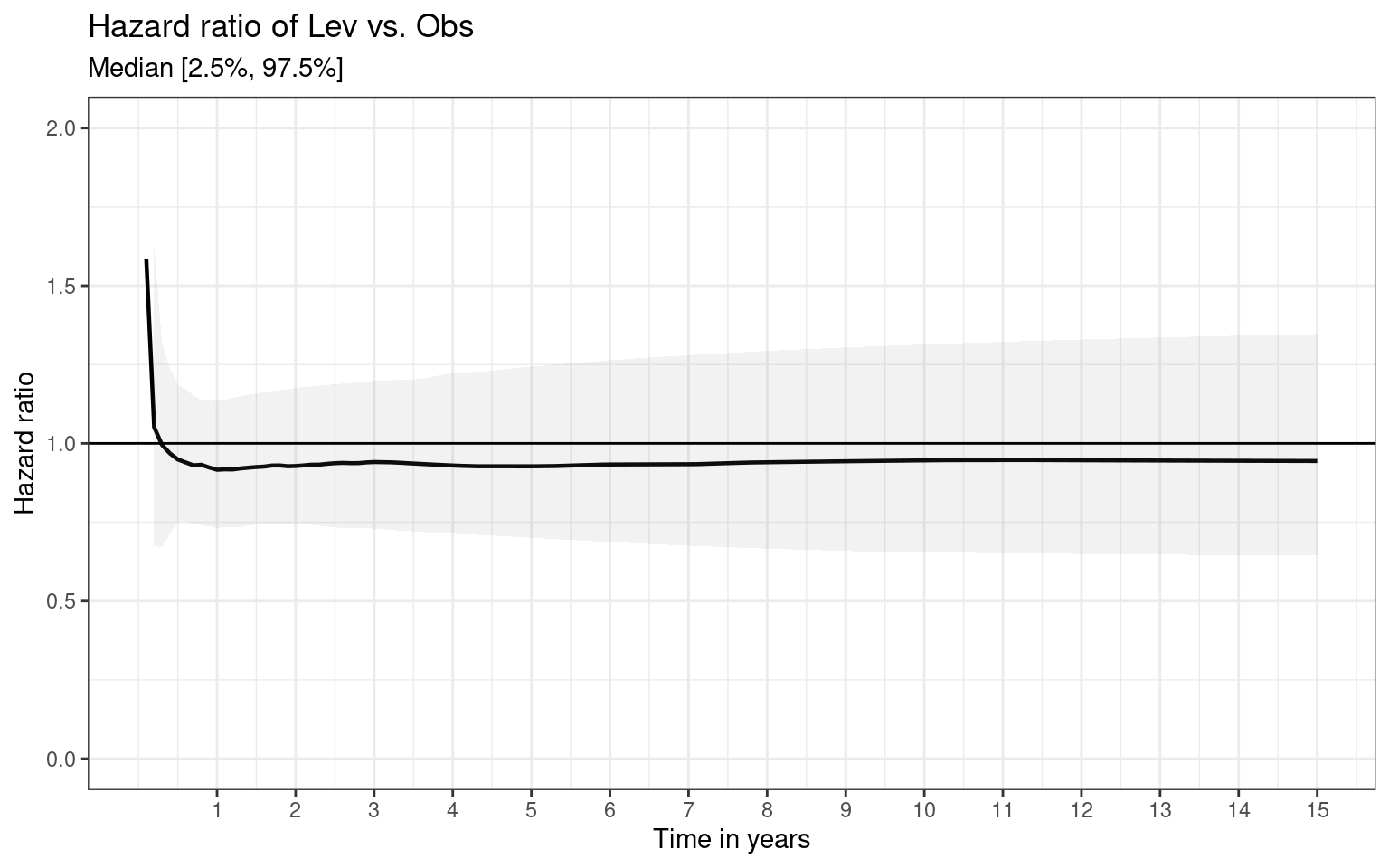
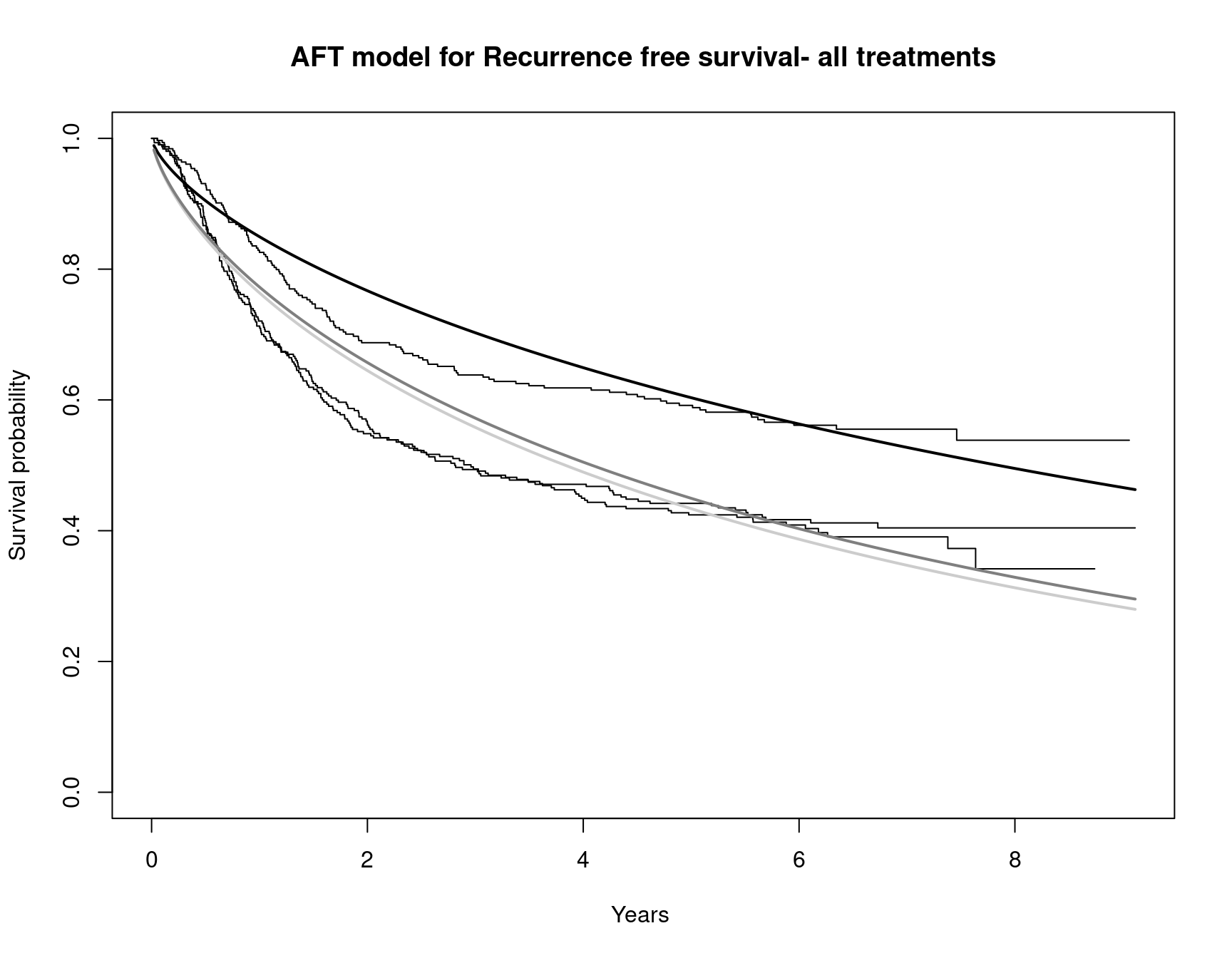
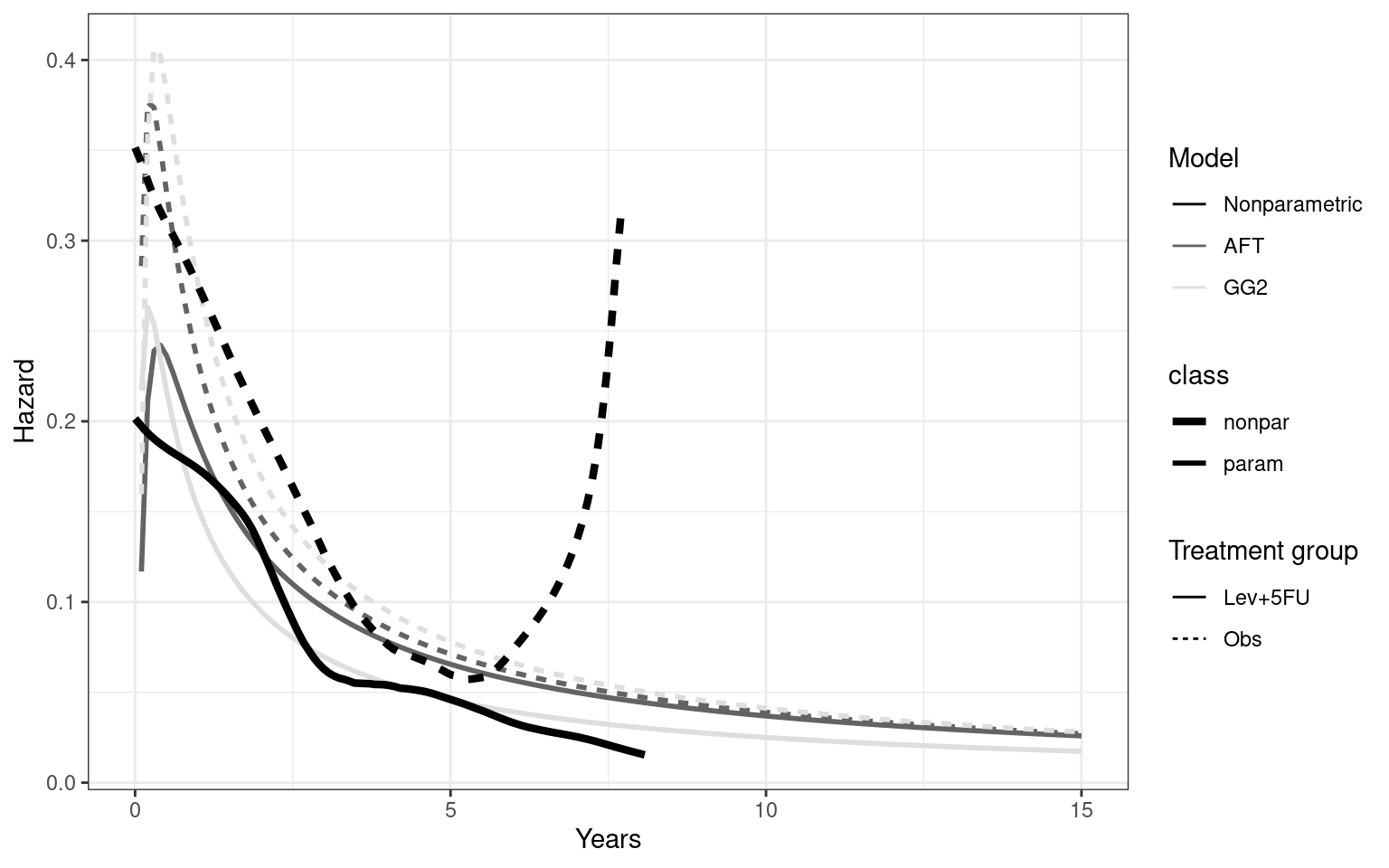
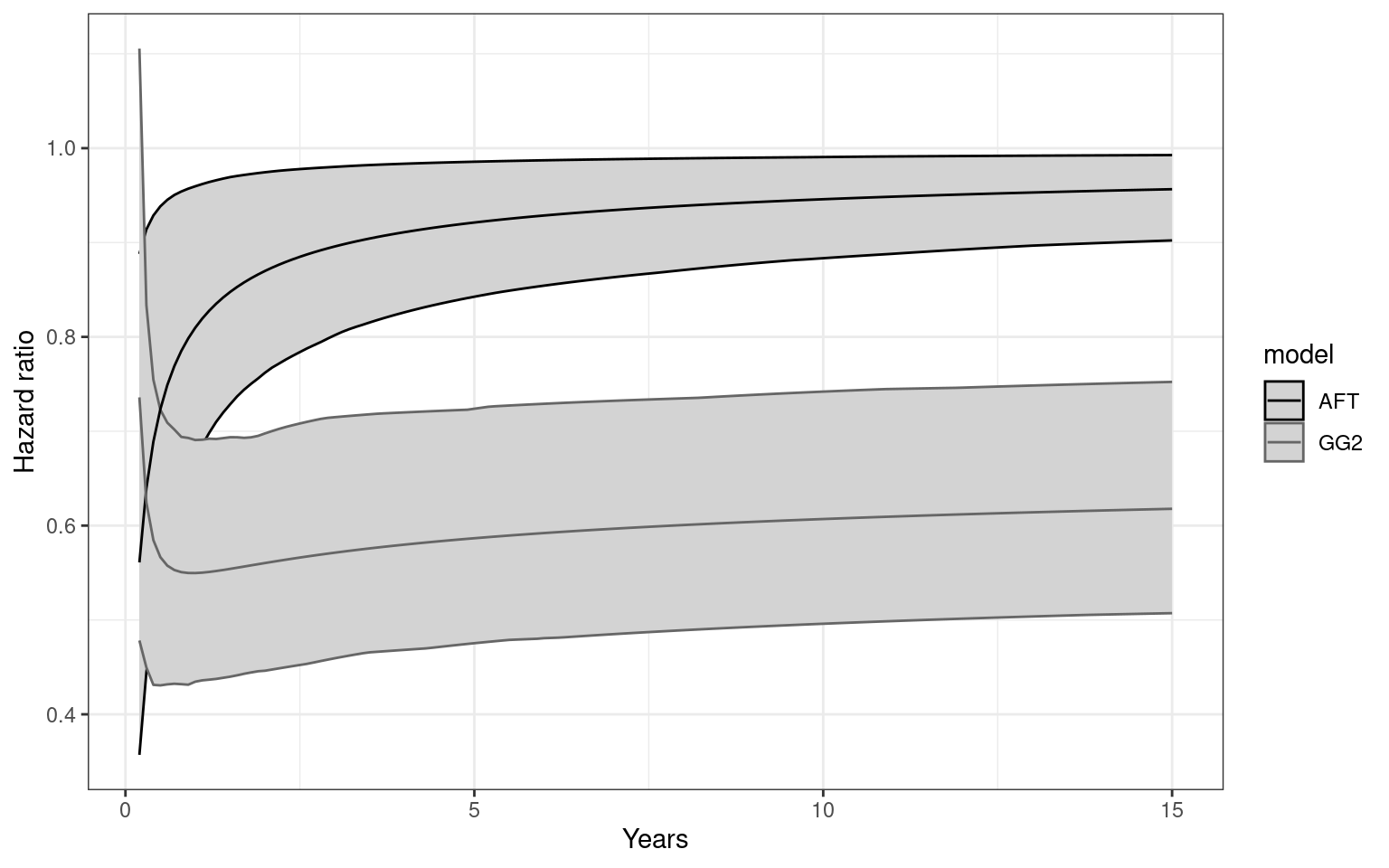
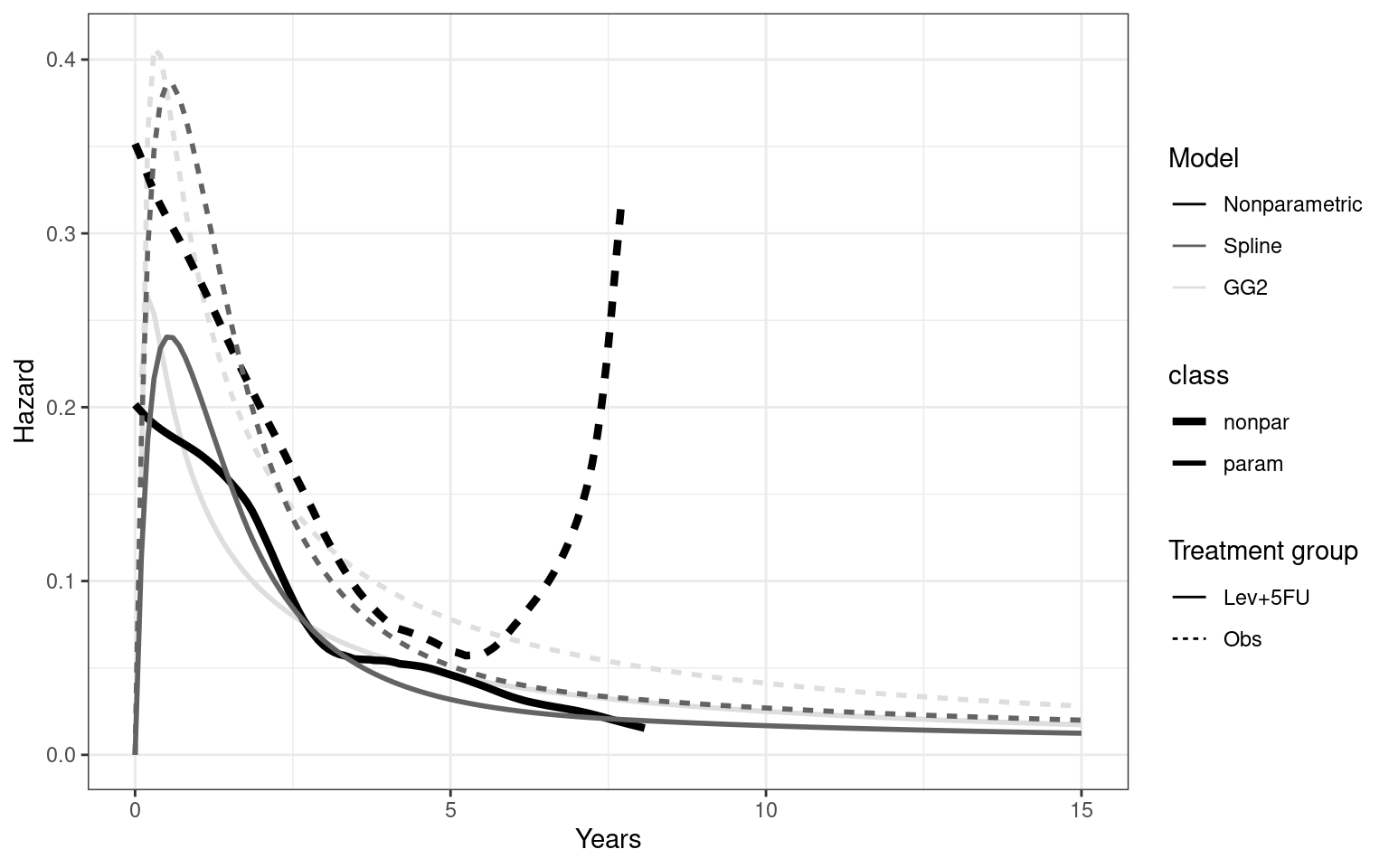
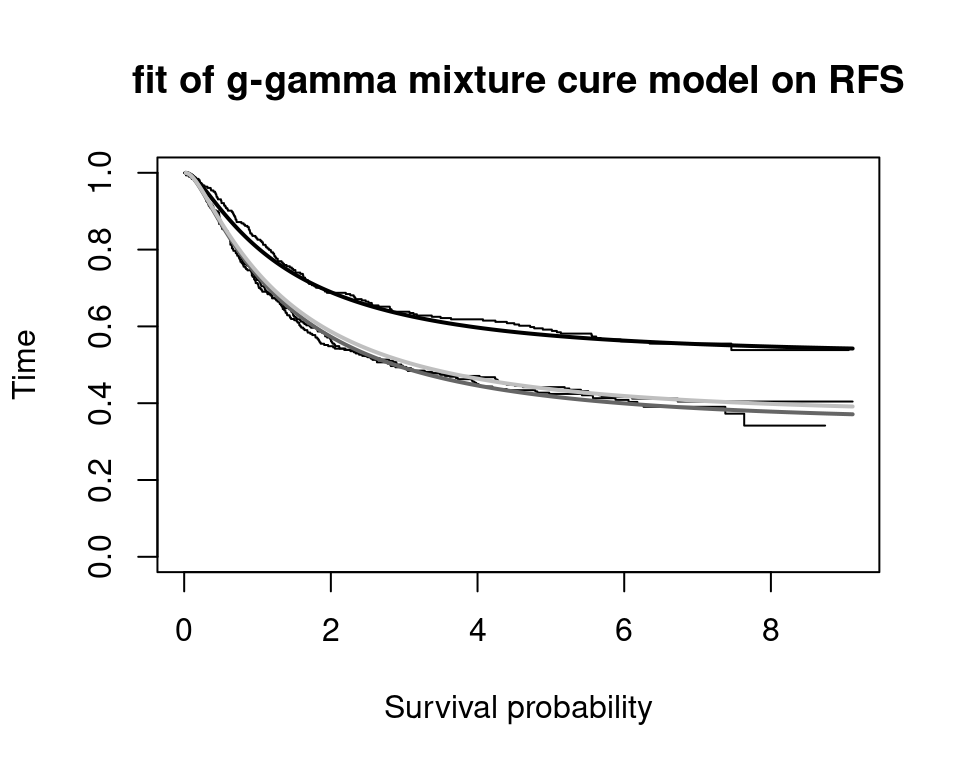
Partitioned survival models (PSM) are types of decision models that are frequently used in oncology to allocate a hypothetical cohort across distinct health states (usually pre-progression, post-progression and dead states) using information from two survival curves (progression-free survival and overall survival). The outcome underlying a progression-free survival (PFS) curve is a composite outcome of either the event of progression or death. Because of this outcome definition, the PFS probability essentially defines the proportion of the cohort, for each time \(t\), that remains in the progression-free state. Subsequently, the complement of the overall survival (OS) probability defines the proportion of the cohort that, at each time, of occupying the dead state. Once the proportion of occupying the progression-free and dead health states are defined, it is straightforward to estimate the proportion of the cohort occupying the progressed state, assuming only these three states exist and this is a closed cohort of individuals.
The PSM approach works effectively in estimating health state occupancy when the sample has been followed fully throughout the decision model’s time horizon. However, when the follow-up time in the studies that inform the OS and PFS probabilities are considerably less than the time horizon of the decision model, extrapolations are necessary for the OS and PFS probabilities. This is most often achieved through fitting parametric survival models to OS and PFS data using methods similar to those described above. However, given that OS and PFS are not independently defined, extrapolations from these parametric survival models and their subsequent application in PSMs can yield biased extrapolations. An extreme example of such inconsistency is when extrapolated PFS probabilities lie above the extrapolated OS probabilities, implicitly implying that a negative proportion of the cohort is occupying the progressed state. Therefore, modellers need to be careful in their considerations of the use of PSMs in decision modelling, especially when the data follow-up time is considerably smaller than the time horizon and/or when few events of the terminal health states (e..g death) have been observed. There is good documentation in the literature on the limitations of PSMs and the advantages of using state-transition and multi-state modelling as alternatives.
Achana, F.A., Cooper, N.J., Bujkiewicz, S., Hubbard, S.J., Kendrick, D., Jones, D.R., Sutton, A.J., 2014. Network meta-analysis of multiple outcome measures accounting for borrowing of information across outcomes. BMC Medical Research Methodology 14.
https://doi.org/10.1186/1471-2288-14-92
Ades, A.E., 2003. A chain of evidence with mixed comparisons: models for multi
-parameter synthesis and consistency of evidence. Statistics in Medicine 22, 2995–3016.
https://doi.org/10.1002/sim.1566
Baio, G., 2020. survHE: survival analysis for health economic evaluation and cost-effectiveness modeling. Journal of Statistical Software 95, 1–47.
Bucher, H.C., Guyatt, G.H., Griffith, L.E., Walter, S.D., 1997. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. Journal of Clinical Epidemiology 50, 683–91.
https://doi.org/10.1016/s0895-4356(97)00049-8
Che, Z., Green, N., Baio, G., 2023. Blended survival curves: A new approach to extrapolation for time-to-event outcomes from clinical trials in health technology assessment. Medical Decision Making 43, 299–310.
Cope, S., Chan, K., Jansen, J.P., 2020. Multivariate network meta-analysis of survival function parameters. Research Synthesis Methods 11, 443–456.
https://doi.org/10.1002/jrsm.1405
Crowther, M.J., Lambert, P.C., 2014. A general framework for parametric survival analysis. Stat. Med. 33, 5280–5297.
Dias, S., Sutton, A.J., Ades, A.E., Welton, N.J., 2013. Evidence synthesis for decision making 2: A generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Medical Decision Making 33, 607–17.
https://doi.org/10.1177/0272989X12458724
Dziak, J.J., Coffman, D.L., Lanza, S.T., Li, R., Jermiin, L.S., 2019. Sensitivity and specificity of information criteria. Briefings in Bioinformatics 21, 553–565.
https://doi.org/10.1093/bib/bbz016
Grafféo, N., Latouche, A., Le Tourneau, C., Chevret, S., 2019. ipcwswitch: An R package for inverse probability of censoring weighting with an application to switches in clinical trials. Computers in Biology and Medicine 111, 103339.
https://doi.org/10.1016/j.compbiomed.2019.103339
Guyot, P., Ades, A., Ouwens, M.J., Welton, N.J., 2012. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Medical Research Methodology 12.
https://doi.org/10.1186/1471-2288-12-9
Hess, S. original by K., Gentleman, R. port by R., 2021.
Muhaz: Hazard function estimation in survival analysis.
Hutton, B., Salanti, G., Caldwell, D.M., Chaimani, A., Schmid, C.H., Cameron, C., Ioannidis, J.P.A., Straus, S., Thorlund, K., Jansen, J.P., Mulrow, C., Catalá-López, F., Gøtzsche, P.C., Dickersin, K., Boutron, I., Altman, D.G., Moher, D., 2015. The PRISMA Extension Statement for Reporting of Systematic Reviews Incorporating Network Meta-analyses of Health Care Interventions: Checklist and Explanations. Annals of Internal Medicine 162, 777–784.
https://doi.org/10.7326/m14-2385
Jackson, C., 2016.
flexsurv: A Platform for Parametric Survival Modeling in R. Journal of Statistical Software 70.
https://doi.org/10.18637/jss.v070.i08
Jackson, C., Stevens, J., Ren, S., Latimer, N., Bojke, L., Manca, A., Sharples, L., 2016. Extrapolating Survival from Randomized Trials Using External Data: A Review of Methods. Medical Decision Making 37, 377–390.
https://doi.org/10.1177/0272989x16639900
Jansen, J.P., 2012. Network meta-analysis of individual and aggregate level data. Research Synthesis Methods 3, 177–90.
https://doi.org/10.1002/jrsm.1048
Jansen, J.P., 2011. Network meta-analysis of survival data with fractional polynomials. BMC Medical Research Methodology 11, 61.
https://doi.org/10.1186/1471-2288-11-61
Jansen, J.P., Fleurence, R., Devine, B., Itzler, R., Barrett, A., Hawkins, N., Lee, K., Boersma, C., Annemans, L., Cappelleri, J.C., 2011. Interpreting indirect treatment comparisons and network meta-analysis for health-care decision making: Report of the ISPOR task force on indirect treatment comparisons good research practices: Part 1. Value Health 14, 417–28.
https://doi.org/10.1016/j.jval.2011.04.002
Lambert, P.C., Thompson, J.R., Weston, C.L., Dickman, P.W., 2007. Estimating and modeling the cure fraction in population-based cancer survival analysis. Biostatistics 8, 576–594.
Latimer, N.R., 2013. Survival analysis for economic evaluations alongside clinical trials–extrapolation with patient-level data: Inconsistencies, limitations, and a practical guide. Med. Decis. Making 33, 743–754.
Latimer, N.R., Rutherford, M.J., 2024. Mixture and non-mixture cure models for health technology assessment: What you need to know. Pharmacoeconomics 42, 1073–1090.
Le Rest, K., Pinaud, D., Monestiez, P., Chadoeuf, J., Bretagnolle, V., 2014. Spatial leave
-one
-out cross
-validation for variable selection in the presence of spatial autocorrelation. Global Ecology and Biogeography 23, 811–820.
https://doi.org/10.1111/geb.12161
Liu, Xing-Rong, Pawitan, Y., Clements, M., 2018. Parametric and penalized generalized survival models. Stat. Methods Med. Res. 27, 1531–1546.
Liu, X.R., Pawitan, Y., Clements, M., 2018. Parametric and penalized generalized survival models 27, 1531–1546.
Moertel, C.G., Fleming, T.R., Macdonald, J.S., Haller, D.G., Laurie, J.A., Tangen, C.M., Ungerleider, J.S., Emerson, W.A., Tormey, D.C., Glick, J.H., Veeder, M.H., Mailliard, J.A., 1995.
Fluorouracil plus levamisole as effective adjuvant therapy after resection of stage III colon carcinoma: a final report. Annals of Internal Medicine 122, 321–6.
https://doi.org/10.7326/0003-4819-122-5-199503010-00001
Ouwens, M.J.N.M., Philips, Z., Jansen, J.P., 2010. Network meta
-analysis of parametric survival curves. Research Synthesis Methods 1, 258–271.
https://doi.org/10.1002/jrsm.25
Prentice, R., 1975. Discrimination among some parametric models. Biometrika 62, 607–614.
Royston, P., Parmar, M.K., 2013. Restricted mean survival time: An alternative to the hazard ratio for the design and analysis of randomized trials with a time-to-event outcome. BMC medical research methodology 13, 1–15.
Zhang, J.Z., Rios, J.D., Pechlivanoglou, T., Yang, A., Zhang, Q., Deris, D., Cromwell, I., Pechlivanoglou, P., 2024. SurvdigitizeR: an algorithm for automated survival curve digitization. BMC Medical Research Methodology 24.
https://doi.org/10.1186/s12874-024-02273-8